



一、基本情况
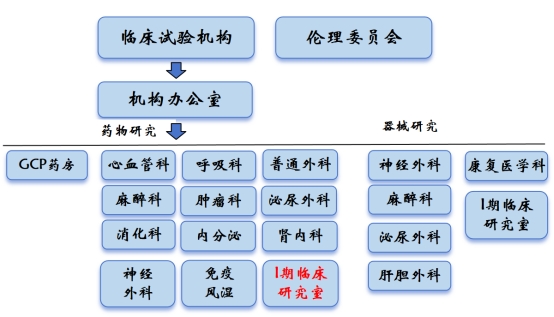
手机彩票2009年经国家药品监督管理局(NMPA)审核、批准,成为国家药物临床试验机构(以下简称“机构”),获准心血管、神经内科、泌尿外科、神经外科、普通外科5个试验专业资质,并于2012年通过复评审查。2014年经NMPA现场资格认定检查,再次获准肿瘤、呼吸、麻醉、消化4个新试验专业资质;2017年9个专业均通过复评审查;2018年6月5日医疗器械临床试验机构完成首次备案,2020年07月药物临床试验机构完成首次机构备案,目前药物临床试验备案了12个专业:心血管、泌尿、普通外科、神经外科、呼吸、麻醉、消化、肿瘤、 I期临床研究室、内分泌、肾内及风湿免疫专业。目前医疗器械临床试验备案了6个专业:神经外科、泌尿、肝胆外科、麻醉科、康复医学科、I期临床研究室。
手机彩票Ⅰ期临床研究室(简称“Ⅰ期临床研究室”)于2016年10月17日成立并正式运行,目前总面积约2416m2,共有76张床位。研究室将各功能区域进行了分隔,办公区、体检区、试验区、生活区相互独立,配有研究病房、受试者活动区、餐厅、集中采样区、样本处理保存室、 尿样收集处理室、吸入给药室、I期药房、监查员办公室等,全区域指纹门禁覆盖。为了确保受试者的安全,研究室设独立的抢救室,配有抢救床、除颤器、呼吸机、心电监护仪、紧急转运车等各种急救设备,同时每个病房配备床头设备带、紧急呼叫系统。为了保证试验顺利进行,研究室配有同步时钟、温控系统、门禁系统。
Ⅰ期临床研究室有独立的质量部和临床研究部两个部门,质量部现有独立专职QA3名,负责整个研究室的体系管理及文件受控。临床研究部现有专职人员21人,包括项目管理人员、研究医生、研究护士、研究药师、QC,其中独立的QC专职负责临床试验的质量控制,专业的人员配备更好的保证了临床药物试验的质量。I期临床研究室开展的一项生物等效性试验于2022年07月以“零缺陷”通过美国食品药品监督管理局(FDA)的远程核查,成为海南省首家通过FDA检查的临床试验机构。
医院具有完备的临床新药科研基础设施和技术条件,重视质量管理,能够保证试验过程的规范性操作,确保项目顺利实施,并在新药临床研究领域获得业内一致性的好评和高度的认可。
目前,医院已有百余名医护人员接受过临床试验相关法规的正规培训,具备开展国内、国际多中心的临床试验研究经验,已逐步建立成为高水平、高质量且能与国际接轨的药物临床试验研究平台。随着不断完善的管理制度以及管理团队,本机构提供服务、监管、协调、培训等职能的管理模式,竭力为申办者、研究者提供高效的质量管理,保证试验过程的规范,试验结果的真实可靠,保障受试者的权益。
二、机构设置
三、临床试验项目开展的工作流程/办事指南
1. 项目立项
(1) 申办方将临床试验方案发至机构办邮箱,机构办查看项目是否与在研项目重复或类似并反馈至申办方。
(2) 申办方填写《临床试验立项申请表》签名后发送扫描件至机构办邮箱,同时按照《临床试验项目申请资料清单》提交相关资料。在机构办审核材料及申请表后,PI及专业负责人审核资料并在立项申请表中签署意见,确认是否承担项目。
(3) 如PI同意承接项目,将专业负责人已审批同意的立项申请表及PI签署的《无利益冲突声明》,提至机构办审核。
2. 伦理审查
(1) 请参照本院伦理委员会章程准备初审材料。
(2) 将初审材料递交至机构办审核,审核无误后递交至伦理委员会。
3. 签署合同
联系机构办进行合同的洽谈签署等工作。
4. 物资交接
合同签署,首付款后监查员联系机构和GCP药房做好试验用药品及物资的交接并做好记录。
5. 监查员备案
(1) 未在机构进行备案的监查员一律不准在本中心做相关的监查工作。
(2) 参与本中心监查的监查员需在项目开展前携带个人资料(包括公司委托书原件、简历,GCP证书、学历证书复印件等相关资料)至机构办进行备案。
6. 临床研究协调员(CRC)备案
(1) 未在机构进行备案的CRC一律不准参与任何项目工作。
(2) 拟授权CRC人员需在上岗前携带个人资料(包括盖章派遣函及相关资质证明、简历,相关专业学历证明、GCP证书等相关资料)至机构办进行备案。
(3) CRC在机构办公室完成上岗前培训,考核合格后方可进行备案。
(4) 备案后发放工牌并签署保密协议,在工牌有效期内在院内开展临床试验相关工作。
(5) 已授权的CRC人员发生变更,需至少提前两周向机构办报备。
7. 项目启动
(1) 项目启动前根据情况召开项目讨论会,对项目的具体实施进行讨论。
(2) 获得本机构伦理批件、签署合同才可召开启动会。如涉及到人类遗传资源管理的项目,需获得相关遗传办批准后才可召开启动会。
(3) 启动会时,项目相关人员(研究者、研究药师等)对方案进行培训。
8. 文件受控
(1) 临床试验过程中由研究人员填写记录的重要原始数据文件/表格均需申请受控。可使用机构办的文件受控管理方式,如项目组或专业有自行受控管理方式,可按照项目组或专业的受控方式执行。
(2) 如使用机构办的文件受控管理方式,在启动会前研究者与监查员、CRC等共同商讨需要进行受控的文件,由研究医生填写《临床试验文件受控清单》交至机构办进行确认。研究人员根据已确认后的《临床试验文件受控清单》准备好相关文件后提交至机构办进行受控。
9. 项目进行中
(1) 监查员每次来中心监查前需提前与机构办预约,并到机构办登记。
(2) 试验过程中所有的方案违背、临床试验方案和知情同意书的修订情况、SAE报告、小结报告、研究进展报告、安全性报告等资料均需递交机构办。
(3) 申办者/CRO变更或转让需及时告知机构办并按照要求提供相关材料。
(4) 如因任何原因主要研究者需要变更,需由申办者代表提交《主要研究者变更申请表》。
10. 项目结题、归档
(1) 项目结题前需将本中心的款项结清。
(2) 项目小结、总结资料需完成本机构的审核后方可签字盖章。
(3) 确认项目资料完整后,按照《临床试验资料归档目录》的要求联系机构办质控员归档。
11. SAE/SUSAR管理
SAE/SUSAR上报邮箱:hksyysaesusar@163.com
12. 联系方式
联系方式: 0898-66151623 13707574885(韩老师)
邮 箱:hksyygcp@163.com
工作时间:周一至周五上午08:00-12:00,下午14:30-17:30
四、相关下载资料
(1) 临床试验立项申请表
(2) 临床试验项目申请资料清单
(3) 无利益冲突声明
(4) 临床试验文件受控清单
(5) 主要研究者变更申请表
(6) 临床试验资料归档目录